To speed patient access to potentially life-saving therapies, the FDA established expedited approval pathways for selected drugs that fill unmet needs or treat serious conditions. For patients in need, these pathways serve an essential purpose. But many payers and providers are concerned that expedited pathways do not require sufficient safety and efficacy data.
Their concerns impact coverage decisions, reimbursement models, and ultimately, patient access. The four key approval pathways are as follows:
- Fast track: Manufacturers apply for this designation, which is meant to speed the approval of drugs for unmet treatment areas. Fast-tracking features frequent FDA/manufacturer communication throughout drug development and review.
- Breakthrough therapy: A manufacturer can apply for this designation by the end of Phase II if preliminary evidence (from a clinically significant endpoint) indicates that its drug may demonstrate substantial improvement over available therapies.
- Accelerated approval: This process allows for conditional approval based on a surrogate endpoint rather than multiple endpoints, for drugs for serious conditions. Manufacturers must conduct phase IV confirmatory trials to earn final approval.
- Priority review: This FDA categorization for drugs for serious conditions shaves about four months off the standard review timeline. This designation doesn’t alter the medical standard for approval or the quality of evidence necessary.
Many payers are reluctant to cover drugs approved via these expedited processes, due in part to a lack of transparency regarding trial outcomes. For drugs granted conditional approval, payers are also concerned about the potential for revoked approval. As a result, some payers postpone P&T review or impose coverage restrictions until after a confirmatory study is completed.
To better understand payers’ point of view on expedited approvals, the MMIT Indices team recently surveyed a representative sample of payers via one of its monthly Market Event Primers. Let’s look at the data.
Restrictions and New-to-Market Blocks
Almost half (45%) of surveyed payers confirmed that they do place restrictions on expedited drugs, primarily prior authorizations and step therapy restrictions. Although most payers implement such restrictions across all lines of business and TAs, some exempt Medicare and Medicaid plans, while others allow unrestricted coverage for oncology, orphan, and pediatric treatments.
Half of payer respondents sometimes choose to exclude expedited drugs from their formularies for a defined period. However, some payers indicated that receiving clinical and real-world evidence on the efficacy and long-term safety of these drugs might cause them to mitigate the effects of a new-to-market block. According to one payer’s pharmacy director, they look for “robust clinical trial results (efficacy & safety beyond endpoints); RWE, Comparative effectiveness (H2H trials); long-term safety data; guideline inclusion (NCCN, ADA, AHA); and cost-effectiveness data.” One CMO simply noted, “There has to be additional clinical trial results showing the efficacy and safety of the drug.”
For other payers, however, new-to-market blocks are not subject to negotiation. As one pharmacy director said, “We implement a new-to-market block until our P&T can review. No data can remove that block.” Of the four distinct expedited approval pathways, payers are most concerned about drugs granted accelerated approval (AA) and least concerned about drugs selected for priority review (see below graphic).
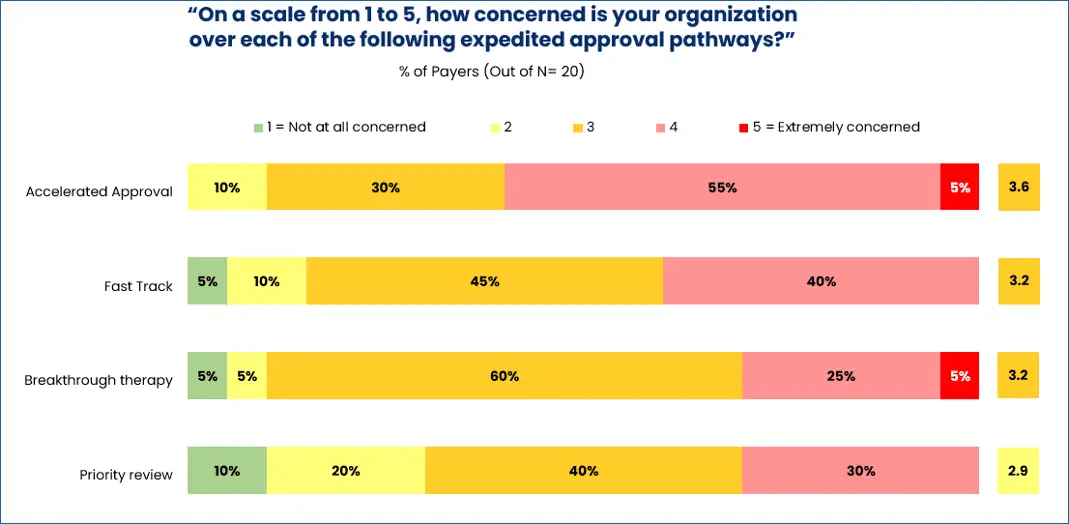
In February, one payer, the Philadelphia-based Independence Blue Cross (IBX), made headlines with its implementation of a new policy to exclude non-oncology AA drugs from coverage for 18 months. Following that period, IBX will re-evaluate its coverage decisions based on the clinical evidence accumulated during that timeframe.
While many pharma companies worried that such extended new-to-market blocks for AA drugs might become commonplace, almost half of surveyed payers said they are unlikely to follow in IBX’s footsteps. As one CMO noted, “Having a ‘blanket policy’ to exclude all drugs approved on the accelerated approval process prevents patients from accessing potentially life-saving drugs. Drugs still should be reviewed on the strength of the clinical literature.”
Surveyed payers generally felt that policies like IBX’s will lead to restricted access, which could negatively influence the prescribing of expedited drugs. “It will obviously be restrictive and create ill will with providers. There will also be legal challenges and regulatory intervention,” said one CMO of a Blues affiliate. A pharmacy director for a large national plan concurred, stating, “There could be significant member and provider abrasion when an FDA approved therapy is not available.”
How Can Manufacturers Address Payer Concerns?
Overall, payers were concerned not only about the lack of data generated through the AA pathway, but also about the rules and requirements for data review. From “higher risk of incomplete clinical validation” to “less stringent data quality requirements,” payers expressed multiple concerns about the “lack of data to review, approved with minimum data requirements or surrogate endpoints.”
So, what can manufacturers of AA drugs do to reassure payers?
1. Educate payers on the validity of surrogate endpoints.
The use of surrogate endpoints, which are easier and faster to measure than clinically meaningful outcomes, is common in AA. If a manufacturer can show that an endpoint correlates with patient outcomes, the FDA is usually willing to accept it as a surrogate.
However, from a payer’s point of view, the strength of association between clinically meaningful outcomes and the surrogates used in trials is often unknown, and therefore suspect. According to many researchers, attempts to validate surrogates are rarely undertaken, and may rely on only a fraction of available data.
The immense backlash against aducanumab (Aduhelm), an AA drug for Alzheimer’s disease, illustrates the pitfalls of relying on surrogate endpoints. While Aduhelm’s approval was based on its ability to reduce amyloid beta plaques, many questions were raised about the validity of amyloid load as a surrogate endpoint in the first place.
From a market access perspective, manufacturers must ensure payers understand the strength of the relationship between any surrogate endpoints and patient outcomes. First, however, they may need to rethink how they approach surrogates altogether. In neuroscience, for example, researcher Brooks Leitner argues that “surrogate endpoints must be tightly linked to the molecular or cellular pathway being targeted.” Selected biomarkers should have compelling, patient-level evidence that they are tied to clinical progression.
2. Keep payers informed about the progression of timely confirmatory trials.
While the AA process requires the completion of confirmatory trials for FDA approval, these post-approval trials are often delayed, with the median time to completion 3.5 years from the date of approval. For payers, this is a period of prolonged uncertainty as to whether the drug will ultimately be proven effective.
Since the FDA created the pathway in 1992, the agency has ultimately withdrawn accelerated approval for 13% of such drugs (41 of 315), as of 2024. Perhaps more importantly for a market access perspective, nearly one-third (32.7%) have not yet been converted to traditional approval and/or have ongoing post-marketing studies.
In January 2025, the FDA issued new draft guidance on what factors it will consider when determining whether a confirmatory trial is underway prior to AA. As it stands now, the FDA will sometimes require participants to be already enrolled in such trials before granting conditional approval.
Manufacturers of drugs expedited through AA must ensure that their confirmatory trials demonstrate clinical benefits as quickly and persuasively as possible. Manufacturers should consider publicly disclosing semi-annual progress reports about their confirmatory trials to demonstrate drug safety and clinical effect, including the number of participants recruited thus far and the estimated time until the trial is complete.
3. Gather (and present) ongoing real-world evidence on cost and efficacy.
According to the results of an AMCP partnership forum, manufacturers can increase payer confidence in AA drugs by systematically collecting real-world data from sources like EHRs, patient registries, and payer databases to “develop confirmatory trial evidence or serve as an external comparator arm for trials.” Such data could also provide manufacturers with measurable benchmarks for morbidity, survival and resource utilization, like the total cost of care, number of emergency visits, cost avoidance, etc.
Through AA, manufacturers have the opportunity to continue collecting real-world data to support their confirmatory trials. Providing transparency into this post-market RWD could help alleviate payer concerns and ease coverage restrictions, including the placement of new-to-market blocks.
Balancing Access and Evidence
To return to the Alzheimer’s space, CMS instituted a coverage with evidence development (CED) policy for all FDA approved early-Alzheimer’s therapies. To receive coverage for traditionally approved Alzheimer’s drugs, Medicare beneficiaries must enroll in a data registry; for AA drugs, they must enroll in clinical studies.
While this policy helps standardize real-world data collection for future and current studies, many experts also believe it unfairly limits treatment access. This conundrum circles back around the essential question for expedited treatment pathways: for patients, is the risk of delaying a potentially life-altering treatment worth the lag time necessary for collecting additional clinical evidence?
The rise of accelerated approvals has magnified tensions between the speed of innovation and the rigor of evidence required for sustainable payer coverage. As the pharma industry continues to evolve, multi-stakeholder collaboration and policy innovation will be critical to ensuring that AA pathways not only deliver rapid access to new therapies, but also maintain public trust.
Manufacturers must be aware that fast-tracking a drug through FDA approval can create new access hurdles for their patients, as it impacts how payers perceive, and ultimately cover, their brands.
Learn how launch analog analysis and forecasting can help your team predict payer coverage with MMIT’s Strategic Launch Report. For more in-depth reporting on access dynamics, consider our Custom Market Research.







